Updated:07/01/2015
BLAST-Basic Local Alignment Tool was born in the 1990s (1,2) and has since been the bread and butter of homology searches (sequence similarity in large databases). Having said that, I would not like to discredit similar tools such as WU-BLAST, FASTA etc. I recently came across and read with great interest a wonderful paper from Baldi and Benz (3) highlighting the possibility of using this tool to fish out similar chemicals from databases. It touches upon a few statistical challenges and adaptations of Tanimoto scores for calculating the similarity of molecules. There are various ways for calculating chemical similarity i.e. graph based, fingerprint based etc. In today’s blog I will discuss using fingerprint based similarity methods to calculate similarity between molecules and how we can use this for BLASTing small molecules. The wiki has a rich page dedicate to the BLAST Tool.
Bringing Chemistry in the BLAST
- Calculate chemical fingerprints (binary: true, false) for a given molecule for size length L.
- Choose the indexes in the L which are set to true (for example, if bits 1, 10, 30,…. etc. are set as ‘true‘).
- Now transform the indexes to char terminated by ‘$’ sign ( i.e 1->B$, 10->BA$, 30->DA$ …)
- Concatenate the patterns to form a sequence (B$BA$DA$…).
- While searching transform them as byte array and match them (treating the byte until $ as one letter).
- Create HSP tuple patterns and align the patterns using the BLAST algorithm.
Here it’s a basic/crude step towards an un-gapped alignment search which is fast yet powerful enough to bring out significant pattern matches between fingerprints sequence arrays.
Performance
In order to test the performances of the method I have chosen ChEMBL-19 dataset containing ~1.4 million molecules.
a) Preparing the ChEMBL-19 database for BLAST search by calculating HSP regions from fingerprints.
Note: This step is only required if a new database is released.
I have used ECFP4 (CDK CircularFingerprint).
Parallel implementation of BLAST search achieves 4X speed improvement, allowing gapped alignment for better coverage of hits.
>sh format.sh data/chembl_19.txt
Formatting input database....
Running Time: 197.04 sec
Index file created chembl_19.idx
DB formatted chembl_19.fmt
b) Performing chemical similarity search on ChEMBL database.
I have chosen the query example selected by Andrew Dalke to test his chemfp-1.2 tool.

“CHEMBL23456″= CNC(=O)c1[nH]c2ccc(Cl)cc2c1Sc1ccccc1
sh search.sh data/chembl_19.txt "CNC(=O)c1[nH]c2ccc(Cl)cc2c1Sc1ccccc1"
Running Time: 70.36 sec, 11808 hits found.
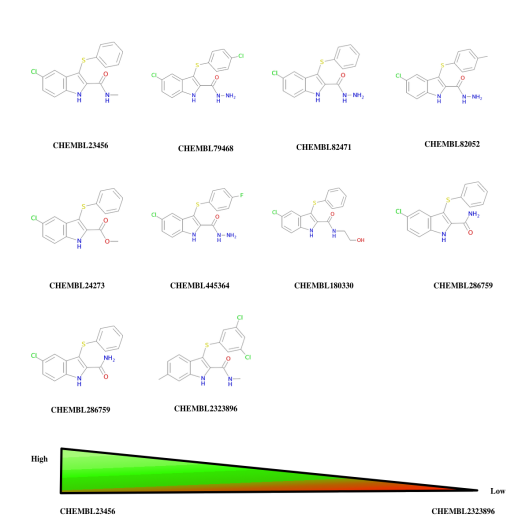
Top 10 ChEMBL-19 hits reported by ChemBLAST tool for query molecule CHEMBL23456.
The top 10 Hits from ChemBLAST tool:
Query Description:
> Query: CNC(=O)c1[nH]c2ccc(Cl)cc2c1Sc1ccccc1
Length = 158
Bit E-
Subject Description Score Value
1> CHEMBL23456 120 4e-26
2> CHEMBL79468 79.7 4e-14
3> CHEMBL82471 77.5 2e-13
4> CHEMBL82052 72.5 7e-12
5> CHEMBL286378 71.7 1e-11
6> CHEMBL24273 69.6 5e-11
7> CHEMBL445364 64.5 2e-09
8> CHEMBL180330 63.7 3e-09
9> CHEMBL286759 62.3 8e-09
10> CHEMBL2323896 62.3 8e-09
For example top two hits are aligned as:
> CHEMBL23456
Length = 158
Score = 120 bits (316), Expect = 4e-26
Identities = 158/158 (100%), Positives = 158/158 (100%), Gaps = 0/158 (0%)
Query 1 BK-KF-BDD-CBG-CCD-CHH-CIB-DHD-DHE-DIG-DKA-ECA-ECC-EEF-EGH-EI 60
BK-KF-BDD-CBG-CCD-CHH-CIB-DHD-DHE-DIG-DKA-ECA-ECC-EEF-EGH-EI
Sbjct 1 BK-KF-BDD-CBG-CCD-CHH-CIB-DHD-DHE-DIG-DKA-ECA-ECC-EEF-EGH-EI 60
Query 61 G-FAF-FAH-FBE-FBF-FCB-FEK-FGK-FHG-GFD-GGB-HBC-HCG-HDH-HEA-HE 120
G-FAF-FAH-FBE-FBF-FCB-FEK-FGK-FHG-GFD-GGB-HBC-HCG-HDH-HEA-HE
Sbjct 61 G-FAF-FAH-FBE-FBF-FCB-FEK-FGK-FHG-GFD-GGB-HBC-HCG-HDH-HEA-HE 120
Query 121 G-HKG-IHA-IKI-KBE-KCG-KEH-KFA-KGH-KKH- 158
G-HKG-IHA-IKI-KBE-KCG-KEH-KFA-KGH-KKH-
Sbjct 121 G-HKG-IHA-IKI-KBE-KCG-KEH-KFA-KGH-KKH- 158
> CHEMBL79468
Length = 154
Score = 79.7 bits (206), Expect = 4e-14
Identities = 129/149 (87%), Positives = 129/149 (87%), Gaps = 8/149 (5%)
Query 6 -BDD-CBG-CCD-CHH-CIB-DHD-DHE-DIG-DKA-ECA-ECC-EEF-EGH-EIG-FAF 65
-B D-CBG-CCD-CHH-CIB-D -DH -D -DKA-E -ECC-EEF-E -EIG-FAF
Sbjct 10 -BHD-CBG-CCD-CHH-CIB-DGB-DHD-DHE-DKA-EAE-ECC-EEF-EIA-EIG-FAF 69
Query 66 -FAH-FBE-FBF-FCB-FEK-FGK-FHG-GFD-GGB-HBC-HCG-HDH-HEA-HE----G 121
-FAH-FBE-FBF-FCB-FE K-FHG-GFD-GGB-HBC-HCG-HDH-HEA-HE G
Sbjct 70 -FAH-FBE-FBF-FCB-FE----K-FHG-GFD-GGB-HBC-HCG-HDH-HEA-HEG-HHG 125
Query 122 -HKG-IHA-IKI-KBE-KCG-KEH-KFA- 150
-HKG-IHA-IKI-KBE-KCG-K H-K A-
Sbjct 126 -HKG-IHA-IKI-KBE-KCG-KGH-KHA- 154
Top 5 hits from Andrew Dalke‘s test case.
1 CHEMBL23456 1.00000
2 CHEMBL180330 0.94105
3 CHEMBL286759 0.93508
4 CHEMBL314618 0.93295
5 CHEMBL382002 0.92812
Thanks to John May, for providing the top 5 chemfp hits using the CDK ECFP4 (makes it an even playing ground as we both use the same fingerprints).
ChEMBL ID Tanimoto Score
1 CHEMBL23456 1.00000
2 CHEMBL82471 0.77778
3 CHEMBL286378 0.74419
4 CHEMBL286759 0.71111
5 CHEMBL24273 0.70213
We all agree with the top hit CHEMBL23456, but we differ on the remaining top 4 hits. This could be due to the variation in choice of our fingerprints/methodology. Nonetheless, the top hits in both the cases looks interesting. I am sure there are many ways to reach the same goal and further optimisation of the code will make it even more attractive. Needs more playing around with!
Suggestions are welcome.
The ChemBLAST– tool is freely available on the GitHub for you to play with and its reasonably fast.
All things come to him who waits – provided he knows what he is waiting for. ~ Woodrow T. Wilson
—————————————————————————————————————————–
Basic principle of sequence BLAST
—————————————————————————————————————————–
To summarise a few of the important concepts:
a) It tries to compare gene sequences (amino acid or nucleotide) – query string against target string. BLAST will find conserved patterns in the database which are similar to sub-patterns in the query.
b) Its uses heuristics by calculating frequently occurring patterns called ‘high-scoring segment pair’ (HSP) in the database (using random walks). It then uses Gumbel extreme value distribution (EVD) to calculate the probability p of observing a score S equal to or greater than x. This is given by the equation:
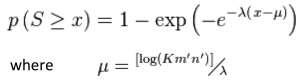
The statistical parameters  and
and  are estimated by fitting the distribution of the un-gapped local alignment scores of the query sequence and a lot of shuffled versions (global or local shuffling) of database sequence patterns, to the Gumbel extreme value distribution.
are estimated by fitting the distribution of the un-gapped local alignment scores of the query sequence and a lot of shuffled versions (global or local shuffling) of database sequence patterns, to the Gumbel extreme value distribution.
c) W: Word/Pattern size – find W-mers (like n-grams) in target/query, T: Threshold – focus on pairs scoring >T, X: Drop-off – stop extending when loss > X, S: Score – the final score of segment pair.
Conditions: If W is too large, it will lead to too many patterns in L and vice versa – if too small, it will lead to too few patterns. If T is too large, then it’s very stringent (conserved blocks) and if too small, it will lead to too many extensions. Look for High-scoring Sequence Pairs (HSPs)-tuples and choose cut-off for relevant hits.
d) Find high scoring pattern of length W, and compile a list L of all W-mers that score >T with some pattern in query sequence. Then scan database for words in L and each positive hit is matched and extended. When score drops more than X below hitherto best score, stop extension. Now report all words with large score S.
References:
1) Altschul SF. et.al. Basic local alignment search tool: J Mol Biol. 1990 Oct 5;215(3):403-10.
2) Altschul SF, et al. Gapped blast and psiblast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402.
3) Baldi P. and Benz RW. BLASTing small molecules—statistics and extreme statistics of chemical similarity scores: Bioinformatics. Jul 1, 2008; 24(13): i357–i365.
52.205337
0.121817

















